Recombinant Human Forkhead box protein G1 (FOXG1)
-
中文名稱:人FOXG1重組蛋白
-
貨號:CSB-YP008815HU
-
規(guī)格:
-
來源:Yeast
-
其他:
-
中文名稱:人FOXG1重組蛋白
-
貨號:CSB-EP008815HU
-
規(guī)格:
-
來源:E.coli
-
其他:
-
中文名稱:人FOXG1重組蛋白
-
貨號:CSB-EP008815HU-B
-
規(guī)格:
-
來源:E.coli
-
共軛:Avi-tag Biotinylated
E. coli biotin ligase (BirA) is highly specific in covalently attaching biotin to the 15 amino acid AviTag peptide. This recombinant protein was biotinylated in vivo by AviTag-BirA technology, which method is BriA catalyzes amide linkage between the biotin and the specific lysine of the AviTag.
-
其他:
-
中文名稱:人FOXG1重組蛋白
-
貨號:CSB-BP008815HU
-
規(guī)格:
-
來源:Baculovirus
-
其他:
-
中文名稱:人FOXG1重組蛋白
-
貨號:CSB-MP008815HU
-
規(guī)格:
-
來源:Mammalian cell
-
其他:
產(chǎn)品詳情
-
純度:>85% (SDS-PAGE)
-
基因名:
-
Uniprot No.:
-
別名:BF-1; BF-2; BF1; BF2; Brain factor 1; Brain factor 2; FHKL; FKH2; FKHL1; FKHL2; FKHL3; FKHL4; Forkhead box protein G1; Forkhead box protein G1A; Forkhead box protein G1B; Forkhead box protein G1C; Forkhead like 1; Forkhead like 2; Forkhead like 3; Forkhead like 4; Forkhead-related protein FKHL1; Forkhead-related protein FKHL2; Forkhead-related protein FKHL3; FOXG1; FOXG1_HUMAN; FOXG1A; FOXG1B; FOXG1C; HBF 1; HBF G2; hBF-2; HBF2; HFK1; HFK2; HFK3; KHL2; Oncogene QIN; QIN
-
種屬:Homo sapiens (Human)
-
蛋白長度:Full length protein
-
表達區(qū)域:1-489
-
氨基酸序列MLDMGDRKEV KMIPKSSFSI NSLVPEAVQN DNHHASHGHH NSHHPQHHHH HHHHHHHPPP PAPQPPPPPQ QQQPPPPPPP APQPPQTRGA PAADDDKGPQ QLLLPPPPPP PPAAALDGAK ADGLGGKGEP GGGPGELAPV GPDEKEKGAG AGGEEKKGAG EGGKDGEGGK EGEKKNGKYE KPPFSYNALI MMAIRQSPEK RLTLNGIYEF IMKNFPYYRE NKQGWQNSIR HNLSLNKCFV KVPRHYDDPG KGNYWMLDPS SDDVFIGGTT GKLRRRSTTS RAKLAFKRGA RLTSTGLTFM DRAGSLYWPM SPFLSLHHPR ASSTLSYNGT TSAYPSHPMP YSSVLTQNSL GNNHSFSTAN GLSVDRLVNG EIPYATHHLT AAALAASVPC GLSVPCSGTY SLNPCSVNLL AGQTSYFFPH VPHPSMTSQS STSMSARAAS SSTSPQAPST LPCESLRPSL PSFTTGLSGG LSDYFTHQNQ GSSSNPLIH
-
蛋白標簽:Tag?type?will?be?determined?during?the?manufacturing?process.
The tag type will be determined during production process. If you have specified tag type, please tell us and we will develop the specified tag preferentially. -
產(chǎn)品提供形式:Lyophilized powder
Note: We will preferentially ship the format that we have in stock, however, if you have any special requirement for the format, please remark your requirement when placing the order, we will prepare according to your demand. -
復溶:We recommend that this vial be briefly centrifuged prior to opening to bring the contents to the bottom. Please reconstitute protein in deionized sterile water to a concentration of 0.1-1.0 mg/mL.We recommend to add 5-50% of glycerol (final concentration) and aliquot for long-term storage at -20℃/-80℃. Our default final concentration of glycerol is 50%. Customers could use it as reference.
-
儲存條件:Store at -20°C/-80°C upon receipt, aliquoting is necessary for mutiple use. Avoid repeated freeze-thaw cycles.
-
保質期:The shelf life is related to many factors, storage state, buffer ingredients, storage temperature and the stability of the protein itself.
Generally, the shelf life of liquid form is 6 months at -20°C/-80°C. The shelf life of lyophilized form is 12 months at -20°C/-80°C. -
貨期:Delivery time may differ from different purchasing way or location, please kindly consult your local distributors for specific delivery time.Note: All of our proteins are default shipped with normal blue ice packs, if you request to ship with dry ice, please communicate with us in advance and extra fees will be charged.
-
注意事項:Repeated freezing and thawing is not recommended. Store working aliquots at 4°C for up to one week.
-
Datasheet :Please contact us to get it.
產(chǎn)品評價

相關產(chǎn)品
靶點詳情
-
功能:Transcription repression factor which plays an important role in the establishment of the regional subdivision of the developing brain and in the development of the telencephalon.
-
基因功能參考文獻:
- Loss of regulatory elements within the refined critical region is the main cause of the FOXG1 syndrome in patients with structural rearrangements associated with long-range position effects. PMID: 29289958
- Patients with FOXG1 syndrome/congenital variant of Rett syndrome associated with pathogenic and likely pathogenic variants in the forkhead box G1 gene indicates substantial variability in overall severity of the phenotype. PMID: 28661489
- We report three cases of FOXG1-related syndrome...with three different underlying genotypes (14q12 deletion including the FOXG1 gene, FOXG1 intragenic mutation, 14q12 deletion including PRKD1 and a region regulating FOXG1 expression) PMID: 29396177
- A novel missense mutation was identified in FOXG1 on gene analysis (c. 569T>A, p. Ile190Asn). The patient showed not only the typical cerebral abnormalities of a congenital variant of Rett syndrome , but also a hypoplastic hippocampus. This novel mutation and cerebral findings may provide new insights into the pathophysiology of the congenital variant of Rett syndrome PMID: 28781028
- The genetic etiology of Rett syndrome (RTT) without MECP2, CDKL5, and FOXG1 mutations is heterogeneous, overlaps with other NDDs, and complicated by a high mutation burden. Dysregulation of chromatin structure and abnormal excitatory synaptic signaling may form two common pathological bases of RTT. PMID: 27171548
- FOXG1 and SOX2 operate in complementary but distinct roles to fuel unconstrained self-renewal in Glioblastoma multiforme stem cells via transcriptional control of core cell cycle and epigenetic regulators. PMID: 28465359
- phenotypes associated with FOXG1 mutations in Chinese Rett syndrome or Rett syndrome-like patients. PMID: 28851325
- describe the initial design and characterizations of novel covalent BH3-based agents that potently target Bfl-1 PMID: 28026162
- findings demonstrate clear phenotype differences between FOXG1 and MECP2 disorders. PMID: 27640358
- Abnormal involuntary movements are a major feature of FOXG1 mutations. Our study delineates the spectrum of movement disorders and confirms an expanding clinical phenotype. Symptomatic treatment may be considered for severe or disabling cases, although further research regarding potential treatment strategies is necessary. PMID: 27029630
- Report demonstrates the functional consequences of Foxg1 haploinsufficiency in the visual system of Foxg1+/Cre mice and a visual impairment in a cohort of Rett individuals presenting genetic alteration on FOXG1 PMID: 27001178
- Upregulated miR-200b in cervical cancer was proven to show positive regulation on cervical cancer development by directly targeting FoxG1. PMID: 27044840
- Rett syndrome with early epilepsy and the congenital variant are mainly due to variations in the CDKL5 and FOXG1 genes, respectively PMID: 26239053
- FOXG1 mutations are associated with familial recurrence in FOXG1-related disorders. PMID: 26364767
- these results implicate the overexpression of a group of neuropeptides in the basal ganglia, hypothalamus, cortex and hippocampus in the pathogenesis FOXG1 behavioral impairments. PMID: 25966633
- These findings suggest a central AKT-FOXG1-reelin signaling pathway in focal malformations of cortical development and support pathway inhibitors as potential treatments or therapies for some forms of focal epilepsy. PMID: 26523971
- We propose that the disruption of signaling pathways that promote mature neuronal differentiation by overexpressed FOXG1 is a contributing event in the neoplastic transformation of cerebellar stem cells. PMID: 26433703
- EGFR mutations remodel the activated enhancer landscape of glioblastoma multiforme, promoting tumorigenesis through a SOX9 and FOXG1-dependent transcriptional regulatory network in vitro and in vivo. PMID: 26455392
- Data suggest that a shift toward GABAergic neuron fate caused by FOXG1 is a developmental precursor of autism spectrum disorder. PMID: 26186191
- The neurological phenotype of FOXG1 haploinsufficiency shows the features of a dyskinetic encephalopathy of infancy. PMID: 25565401
- critical role in the regulation of hepatocellular carcinoma development PMID: 25251503
- Genotype-phenotype studies of FOXG1 may help to elucidate why children develop different forms of developmental epilepsy. PMID: 24836831
- Reduced FOXG1 levels in patients' platelets having translocations or deletions in that region. PMID: 23632790
- transcriptional programmes regulated by FOXG1 and Groucho/TLE are important for BTIC-initiated brain tumour growth, implicating FOXG1 and Groucho/TLE in GBM tumourigenesis PMID: 24356439
- Our data and review of previous reports highlight dysregulation of FOXG1 pathway as the cause of the "FOXG1 syndrome" developmental disorder PMID: 23956198
- Its mutation causes Rett syndrome.(review) PMID: 24738188
- Authors assessed the functional relevance of two genes, FoxG1 and Bmi1, which were significantly enriched in non-Shh/Wnt MBs and showed these genes to mediate MB stem cell self-renewal and tumor initiation in mice. PMID: 23592496
- FoxG1 can function as a pro-apoptotic factor in part through suppression of AIB1 coactivator transcription complex formation, thereby reducing the expression of the AIB1 oncogene. PMID: 23660594
- 14q12 microdeletions excluding FOXG1, but leading to its misregulation give rise to a congenital variant Rett syndrome-like phenotype. PMID: 22968132
- In fibroblast cells, a cis-acting regulatory sequence located more than 0.6 Mb away from FOXG1 acts as a silencer at the transcriptional level. PMID: 22739344
- FOXG1 mutations are involved in the molecular etiology of the congenital variant of Rett syndrome. PMID: 22129046
- Alterations in the kinetics of FoxG1 binding to chromatin might contribute to the pathological effects of FOXG1 mutations. PMID: 22091895
- The authors show that deletions including 14q13 result in a recognisable phenotype mainly due to haploinsufficiency of two genes (NKX2-1, PAX9). FOXG1 (on chromosome band 14q12) involvement seems to be the main determinant of phenotype severity. PMID: 22636604
- Foxg1 is critical for dentate gyrus formation, especially during the early postnatal stage. PMID: 22378868
- A small increase in the dosage of FOXG1 could cause infantile spasms. PMID: 21910242
- The core FOXG1 syndrome phenotype consists of postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and corpus callosum hypogenesis. PMID: 21441262
- West syndrome was associated with 14q12 duplications harboring FOXG1 PMID: 21536641
- Transgenic mice lacking microRNAs miR-9-2 and miR-9-3 exhibit multiple defects in their telencephalic structures which may be brought about by dysregulation of Foxg1, Nr2e1, Gsh2, and Meis2 expression. PMID: 21368052
- We report a series of seven cases of patients with FIXG1 gebe duplications in 14q associated with developmental delay/mental retardation and speech delay as predominant features, as well as developmental epilepsy in the majority. PMID: 20736978
- 150 patients affected by postnatal microcephaly, and identified two mutations: the c.326C>T (p.P109L) substitution and the c.730C>T transition, which induces the p.R244C mutation within the DNA-binding forkhead domain. PMID: 21280142
- two de novo mutations (c.1248C>G, p.Y416X and c.460_461dupG, p.E154GfsX300) were identified in two unrelated girls with Rett syndrome PMID: 19806373
- Two different de novo heterozygous FOXG1-truncating mutations were identified. The subject with the p.Trp308X mutation presented with a severe RTT-like neurodevelopmental disorder, while the p.Tyr400X allele was associated with classical RTT symptoms. PMID: 19564653
- these results contribute to the clarification of the phenotype associated with FOXG1, confirming its role in the Rett syndrome spectrum. PMID: 19578037
- BF-1 and PAX9 interact with PLU-1 via a novel conserved sequence motif (Ala-X-Ala-Ala-X-Val-Pro-X4-Val-Pro-X8-Pro, termed the VP motif) PMID: 12657635
- The expression of FOXG1 showed an inverse relationship. FOXG1 copy gain was seen in 55/59 of a validating set of tumors and showed a positive correlation with protein expression representing the first report of FOXG1 dysregulation in medulloblastoma. PMID: 17522785
- FOXG1 is responsible for the congenital variant of Rett syndrome. PMID: 18571142
顯示更多
收起更多
-
相關疾病:Rett syndrome congenital variant (RTTCV)
-
亞細胞定位:Nucleus.
-
組織特異性:Expression is restricted to the neurons of the developing telencephalon.
-
數(shù)據(jù)庫鏈接:
Most popular with customers
-

Recombinant Human Tumor necrosis factor ligand superfamily member 9 (TNFSF9), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-

Recombinant Mouse Retinol-binding protein 4 (Rbp4) (Active)
Express system: Mammalian cell
Species: Mus musculus (Mouse)
-

Recombinant Human IL12B&IL12A Heterodimer Protein (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-

Recombinant Human Transferrin receptor protein 1 (TFRC), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
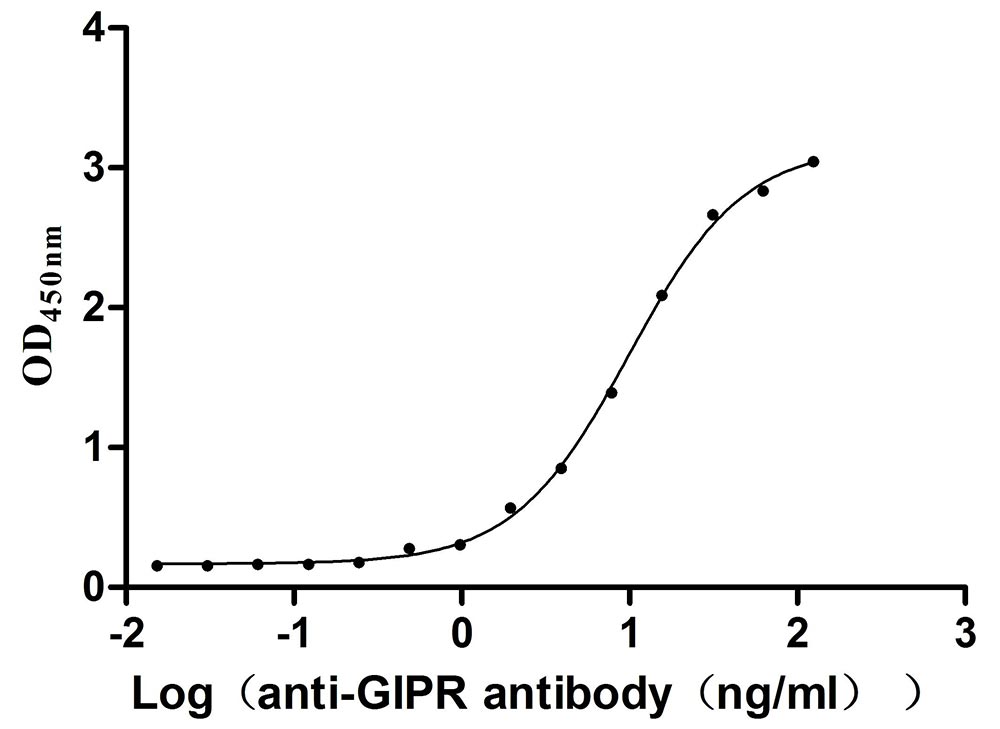
Recombinant Mouse Gastric inhibitory polypeptide receptor (Gipr), partial (Active)
Express system: Mammalian cell
Species: Mus musculus (Mouse)
-

Recombinant Human Carcinoembryonic antigen-related cell adhesion molecule 8(CEACAM8) (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
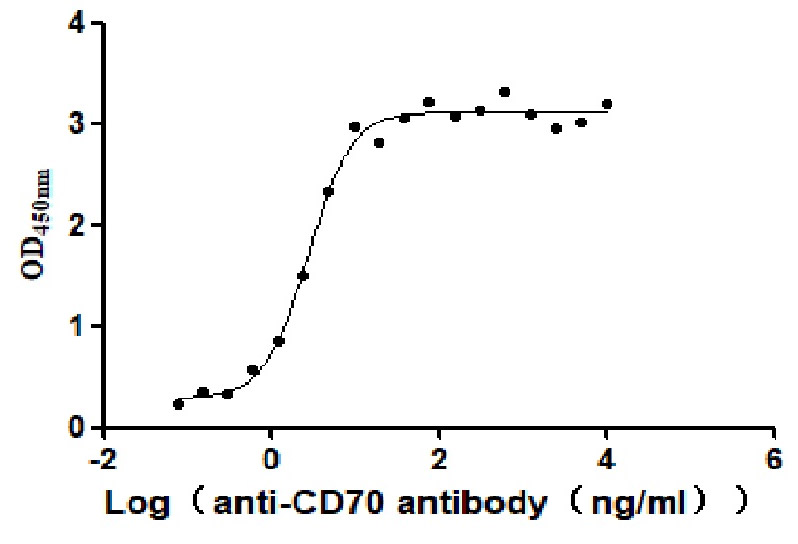
Recombinant Human CD70 antigen (CD70), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-

Recombinant Macaca fascicularis Gastric inhibitory polypeptide receptor (GIPR), partial (Active)
Express system: yeast
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)