Recombinant Human Lysosomal alpha-glucosidase (GAA), partial
-
中文名稱:人GAA重組蛋白
-
貨號(hào):CSB-YP009125HU
-
規(guī)格:
-
來源:Yeast
-
其他:
-
中文名稱:人GAA重組蛋白
-
貨號(hào):CSB-EP009125HU
-
規(guī)格:
-
來源:E.coli
-
其他:
-
中文名稱:人GAA重組蛋白
-
貨號(hào):CSB-EP009125HU-B
-
規(guī)格:
-
來源:E.coli
-
共軛:Avi-tag Biotinylated
E. coli biotin ligase (BirA) is highly specific in covalently attaching biotin to the 15 amino acid AviTag peptide. This recombinant protein was biotinylated in vivo by AviTag-BirA technology, which method is BriA catalyzes amide linkage between the biotin and the specific lysine of the AviTag.
-
其他:
-
中文名稱:人GAA重組蛋白
-
貨號(hào):CSB-BP009125HU
-
規(guī)格:
-
來源:Baculovirus
-
其他:
-
中文名稱:人GAA重組蛋白
-
貨號(hào):CSB-MP009125HU
-
規(guī)格:
-
來源:Mammalian cell
-
其他:
產(chǎn)品詳情
-
純度:>85% (SDS-PAGE)
-
基因名:
-
Uniprot No.:
-
別名:70 kDa lysosomal alpha-glucosidase; Acid alpha glucosidase; Acid maltase; Aglucosidase alfa; Alpha glucosidase; GAA; Glucosidase alpha acid (Pompe disease glycogen storage disease type II); Glucosidase alpha acid; Glucosidase alpha; LYAG; LYAG_HUMAN; Lysosomal alpha glucosidase
-
種屬:Homo sapiens (Human)
-
蛋白長度:Partial
-
蛋白標(biāo)簽:Tag?type?will?be?determined?during?the?manufacturing?process.
The tag type will be determined during production process. If you have specified tag type, please tell us and we will develop the specified tag preferentially. -
產(chǎn)品提供形式:Lyophilized powder
Note: We will preferentially ship the format that we have in stock, however, if you have any special requirement for the format, please remark your requirement when placing the order, we will prepare according to your demand. -
復(fù)溶:We recommend that this vial be briefly centrifuged prior to opening to bring the contents to the bottom. Please reconstitute protein in deionized sterile water to a concentration of 0.1-1.0 mg/mL.We recommend to add 5-50% of glycerol (final concentration) and aliquot for long-term storage at -20℃/-80℃. Our default final concentration of glycerol is 50%. Customers could use it as reference.
-
儲(chǔ)存條件:Store at -20°C/-80°C upon receipt, aliquoting is necessary for mutiple use. Avoid repeated freeze-thaw cycles.
-
保質(zhì)期:The shelf life is related to many factors, storage state, buffer ingredients, storage temperature and the stability of the protein itself.
Generally, the shelf life of liquid form is 6 months at -20°C/-80°C. The shelf life of lyophilized form is 12 months at -20°C/-80°C. -
貨期:Delivery time may differ from different purchasing way or location, please kindly consult your local distributors for specific delivery time.Note: All of our proteins are default shipped with normal blue ice packs, if you request to ship with dry ice, please communicate with us in advance and extra fees will be charged.
-
注意事項(xiàng):Repeated freezing and thawing is not recommended. Store working aliquots at 4°C for up to one week.
-
Datasheet :Please contact us to get it.
產(chǎn)品評(píng)價(jià)

相關(guān)產(chǎn)品
問答及客戶評(píng)論
I'm interested in CSB-MP009125HU and would like to know which mammalian cells are used to express the protein?
靶點(diǎn)詳情
-
功能:Essential for the degradation of glycogen in lysosomes. Has highest activity on alpha-1,4-linked glycosidic linkages, but can also hydrolyze alpha-1,6-linked glucans.
-
基因功能參考文獻(xiàn):
- PI-rhGAA may have the potential to be a useful therapeutic option for improving the treatment of Pompe disease. PMID: 29102549
- The most common mutation was c.-32-13T, G. in Pompe disease. PMID: 29181627
- The narrow substrate-binding pocket of rhGAA is located near the C-terminal ends of beta-strands of the catalytic (beta/alpha)8 domain and shaped by a loop from the N-terminal beta-sheet domain and both inserts I and II. PMID: 29061980
- This is the first study of rhGAA to differentiate M6P glycans and identify their attachment sites, despite rhGAA already being an approved drug for Pompe disease. PMID: 29274340
- GAA mutation is associated with Pompe disease. PMID: 28763149
- Enzyme activities (acid alpha-glucosidase (GAA), galactocerebrosidase (GALC), glucocerebrosidase (GBA), alpha-galactosidase A (GLA), alpha-iduronidase (IDUA) and sphingomyeline phosphodiesterase-1 (SMPD-1)) were measured on ~43,000 de-identified dried blood spot (DBS) punches, and screen positive samples were submitted for DNA sequencing to obtain genotype confirmation of disease risk PMID: 27238910
- enzyme replacement therapy (ERT) (alglucosidase alfa) stabilizes respiratory function and improves mobility and muscle strength in late-onset Pompe disease.Lysosomal glycogen in muscle biopsies from treatment-naive LOPD patients was reduced post-ERT (alglucosidase alfa). PMID: 27473031
- In adults with Pompe disease, antibody formation does not interfere with rhGAA efficacy in the majority of patients, is associated with IARs, and may be attenuated by the IVS1/delex18 GAA genotype PMID: 27362911
- Reanalysis of the patient's DNA sample using next generation sequencing (NGS) of a panel of target genes causing glycogen storage disorders demonstrated compound heterozygosity for a point mutation and an exonic deletion in the GAA gene. PMID: 28657663
- Thirteen novel and two common GAA mutations were identified in this study. The allelic frequency of c.2662G > T (p.Glu888X) was 23.1% in northern Chinese patients and 4.2% in southern Chinese patients, whereas the allelic frequency of c.1935C >A (p.Asp645Glu) was 20.8% in southern and 3.8% in northern Chinese patients. PMID: 28394184
- This is the first report of the alpha-glucosidase inhibitory activity of compounds 20, 26, and 29, and the findings support the important role of Eremanthus species as novel sources of new drugs and/or herbal remedies for treatment of type 2 diabetes. PMID: 27322221
- Compared with controls, GAA gene expression levels in coronary artery disease (CAD) patients were significantly increased, suggesting that GAA may be involved in the CAD development. PMID: 26580301
- Study reports on the clinical, biochemical, morphological, muscle imaging, and genetic findings of six adult Pompe patients from five unrelated families with the c.-32-13T>G GAA gene mutation in homozygous state. All patients had decreased GAA activity and elevated creatine kinase levels. PMID: 26231297
- glycogen storage disease type II is caused by deficiency of GAA activity resulting from mutation of GAA gene PMID: 26575883
- RT-PCR followed by DNA sequence analysis of patients with Pompe disease revealed new variant in GAA gene resulting in aberrant splicing event. PMID: 25243733
- Findings indicate that GAA c.2238G > C (p.W746C) novel mutation is the most common mutation in mainland Chinese late-onset Pompe patients, as observed in Taiwanese patients expanding the genetic spectrum of the disease. PMID: 25526786
- this study shows several alterations distributed along the GAA gene in a sample of Brazilian families. PMID: 25681614
- Mutations in acid alpha-glucosidase gene is associated with Pompe disease. PMID: 25026126
- GAA deficiency results in reduced mTORC1 activation that is partly responsible for the skeletal muscle wasting phenotype and can be amerliorated by leucine supplementation. PMID: 25231351
- The phenotype LO-GSDII with GAA mutation in the North of Italy seems not significantly different from other LO-GSDII populations in Europe or the USA. PMID: 24158270
- Data shows the largest informative family with late-onset Pompe disease described in the literature showing a peculiar complex set of mutations of GAA gene that may partially elucidate the clinical heterogeneity of this family. PMID: 24107549
- 7 of 27 in: Gene. 2014 Mar 1;537(1) Novel GAA sequence variant c.1211 A>G reduces enzyme activity but not protein expression in infantile and adult onset Pompe disease. PMID: 24384324
- This study demonstrates that the c.-32-13T>G mutation of GAA gene abrogates the binding of the splicing factor U2AF65 to the polypyrimidine tract of exon 2 and that several splicing factors affect exon 2 inclusion. PMID: 24150945
- study describes two unrelated cases affected with classical early-onset Pompe disease, both pertaining to the same small Mexican region, with the same novel homozygous frameshift mutation at gene GAA (c.1987delC) PMID: 24399866
- Mutations in the GAA gene is associated with glycogen storage disease type II. PMID: 23884227
- Adult patients with alpha-glucosidase mutations other than c.-32-13 T>G can have very low alpha-glucosidase activity in fibroblasts but express higher activity in muscle and store less glycogen in muscle than patients with infantile Pompe disease. PMID: 23000108
- Study gave an update of the pompe disease mutation database with 60 novel GAA sequence variants and additional studies on the functional effect of 34 previously reported variants. PMID: 22644586
- Transcriptional response to GAA deficiency (Pompe disease) in infantile-onset patients PMID: 22658377
- Report genetic testing to indentify GAA mutations in German patients with late-onset glycogen storage disease type II. PMID: 18607768
- we define a critical role for endoplasmic reticulum stress in the activation of autophagy due to the 546G>T acid alpha glucosidase mutation PMID: 21982629
- No common mutation is found in association with low levels of acid alpha-glucosidase activity in late-onset Pompe disease; most patients produce unprocessed forms of GAA protein compared with patients who have higher GAA activity. PMID: 21484825
- Mutation analysis of the GAA gene revealed the p.D645E in all patients with Pompe disease, suggesting it as the most common mutation in the Thai population. PMID: 21039225
- The enzymatic screening of Pompe disease can be justified in patients with myopathies of unknown etiology in this report of a Mexican patient with late-onset glycogen-storage disease type 2. PMID: 20350966
- Data show that p.R1147G missense mutation impaired glucosidase activity. PMID: 19834502
- Homozygosity for multiple contiguous single-nucleotide polymorphisms as an indicator of large heterozygous deletions: identification of a novel heterozygous 8-kb intragenic deletion (IVS7-19 to IVS15-17) in a patient with glycogen storage disease type II PMID: 11854868
- novel target of the Notch-1/Hes-1 signaling pathway PMID: 12065598
- 2 novel mutations of the acid alpha-glucosidase gene, P361L and R437C, were found in a juvenile-onset glycogen storage disease type II (GSDII) 16-year-old Chinese patient. The asymptomatic 13-year-old brother of the proband is also compound heterozygote PMID: 12601120
- mutations in the alpha glucosidase gene is associated with infantile onset glycogen storage disease type II. PMID: 12923862
- Childhood Pompe disease demonstrating phenotypic variability of p.Asp645Asn. PMID: 15145338
- data show that the mature forms of GAA characterized by polypeptides of 76 or 70 kDa are in fact larger molecular mass multicomponent enzyme complexes; peptides released during proteolytic processing remained tightly associated with the major species PMID: 15520017
- 2 novel mutations (Ala237Val and Gly293Arg) were foundin the acid alpha-glucosidase gene in a Pompe disease patient with vascular involvement. PMID: 15668445
- Acid-alpha-glucosidase activity and specific activity, and lysosomal glycogen content are useful predictors of age of onset in Pompe disease PMID: 15993875
- Complete molecular analysis of the GAA gene of patients with late onset glycogen storage disease type II shows missense mutations and splicing mutations. PMID: 16917947
- From 14 Argentinean patients diagnosed with either infantile or late-onset disease, we identified 14 distinct mutations in the acid alpha-glucosidase (GAA) gene including nine novel variants. PMID: 17056254
- Two new missense mutations (p.266Pro>Ser and p.439Met>Lys) were new missense mutations causing late onset GSD II. PMID: 17092519
- Patients with the same c.-32-13T-->G haplotype (c.q. GAA genotype) may manifest first symptoms at different ages, indicating that secondary factors may substantially influence the clinical course of patients with this mutation. PMID: 17210890
- demonstrated a significant increase of GAA activity (1.3-7.5-fold) after imino sugar treatment in fibroblasts from patients carrying the mutations L552P (three patients) and G549R (one patient) PMID: 17213836
- N-glycans of recombinant human GAA were expressed in the milk of transgenic rabbits. PMID: 17293352
- The role of autophagy in Pompe disease was examined by analyzing single muscle fibers. PMID: 17592248
- Mutations in glucosidase alpha is asspciated with glycogen storage disease type II PMID: 17616415
顯示更多
收起更多
-
相關(guān)疾病:Glycogen storage disease 2 (GSD2)
-
亞細(xì)胞定位:Lysosome. Lysosome membrane.
-
蛋白家族:Glycosyl hydrolase 31 family
-
數(shù)據(jù)庫鏈接:
Most popular with customers
-

Recombinant Human Tumor necrosis factor receptor superfamily member 18 (TNFRSF18), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-

Recombinant Rat Intestinal-type alkaline phosphatase 1 (Alpi) (Active)
Express system: Mammalian cell
Species: Rattus norvegicus (Rat)
-
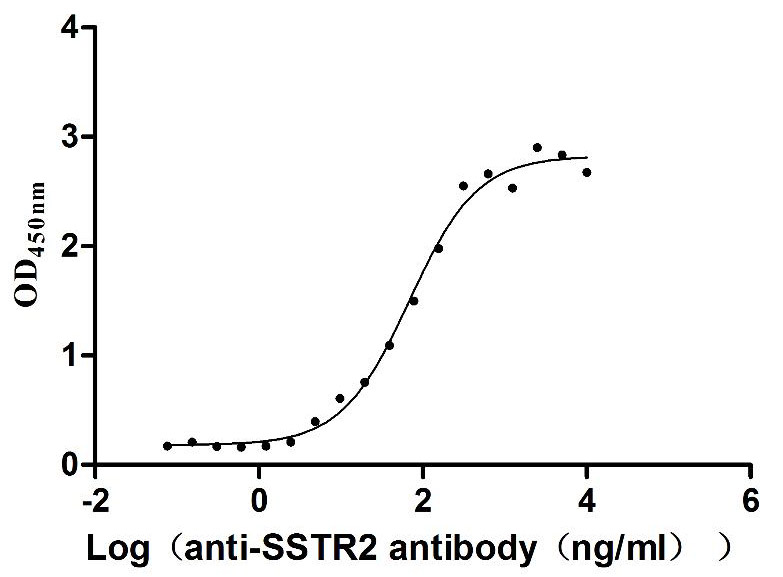
Recombinant Human Somatostatin receptor type 2 (SSTR2)-VLPs (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
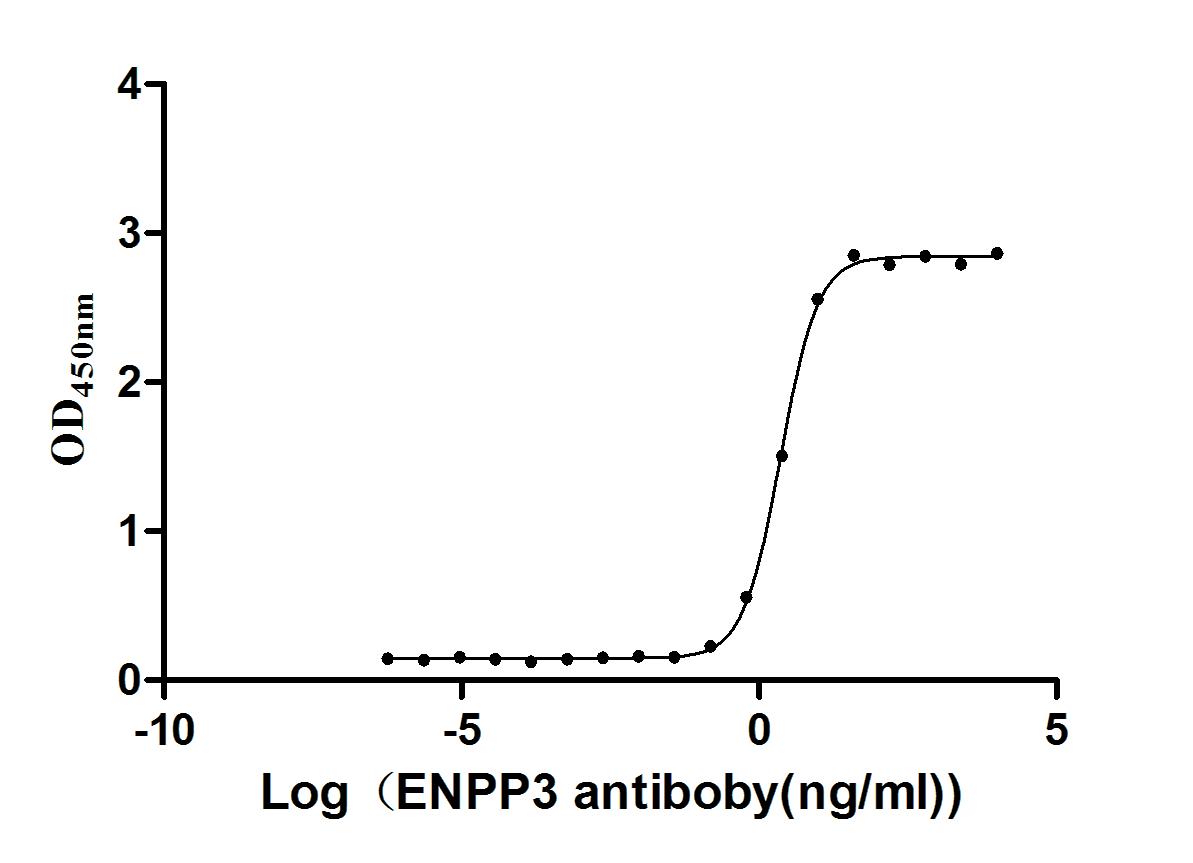
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
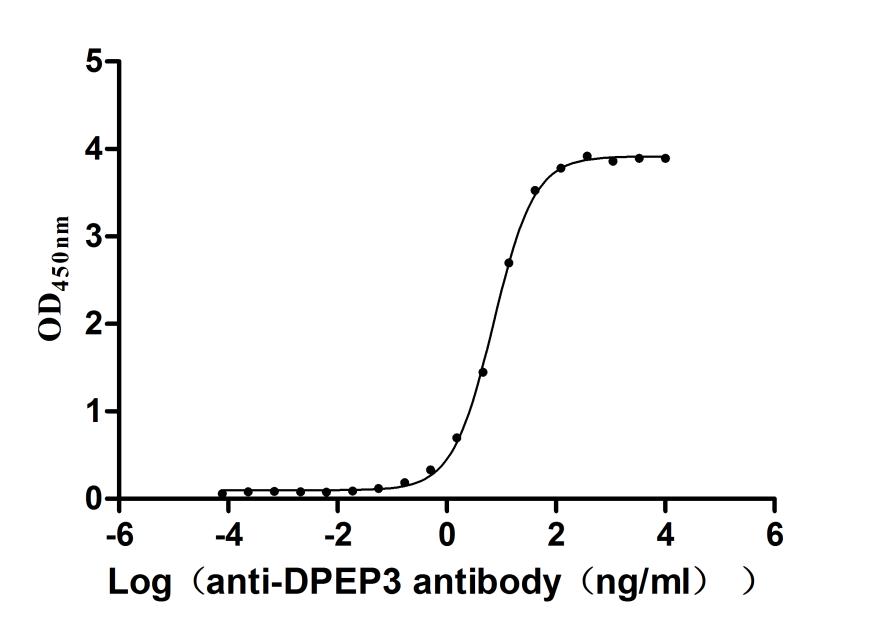
Recombinant Human Dipeptidase 3(DPEP3), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-

Express system: Mammalian cell
Species: Macaca mulatta (Rhesus macaque)